Meet MolXProt: A Fresh Way to Predict How Drugs Bind
What if you could predict how a drug molecules binds to its target protein, while being fast, reliable, and with more chemical interpretability than ever? That’s what MolXProt was designed to achieve.
In the world of drug discovery, predicting the binding affinities between protein and ligand pairs is key. However, traditional methods such as docking and empirical scoring often fail to generalize to novel protein families and new chemistries.
MolXProt was created to give a fresh new look to the problem: A transformer-based graph neural network that fuses graph-based ligand representations with protein language model embeddings via a bidirectional multi-head cross-attention. The results? We were able to obtain a scalable and interpretable model that delivers high-quality predictions and pave the way to understand how drugs and proteins “talk” at the atomic level.
Why it matters?
- Drug discovery bottleneck: Being able to accurately and quickly predict binding affinities is essential. Protein-ligand systems are composed by thousands of atoms, which makes their computational simulation a real challenge.
- Limitations of current methods: As motivated, tradional methods often fail to generalize. On the other hand, a few state-of-the-art ML/AI models are capable of producing chemically accurate results, at the cost of chemical/physical interpretability.
- Our step forward: We combine graph representation of ligands with protein language model embeddings via a cross-attention fusion, allowing the model to directly learn interactions between ligand atoms and protein residues.
- Scalable and efficient: MolXProt is designed to train on large datasets (100000+ protein-ligand pairs), while remaining computationally friendly and trainable on a Macbook Air M4.
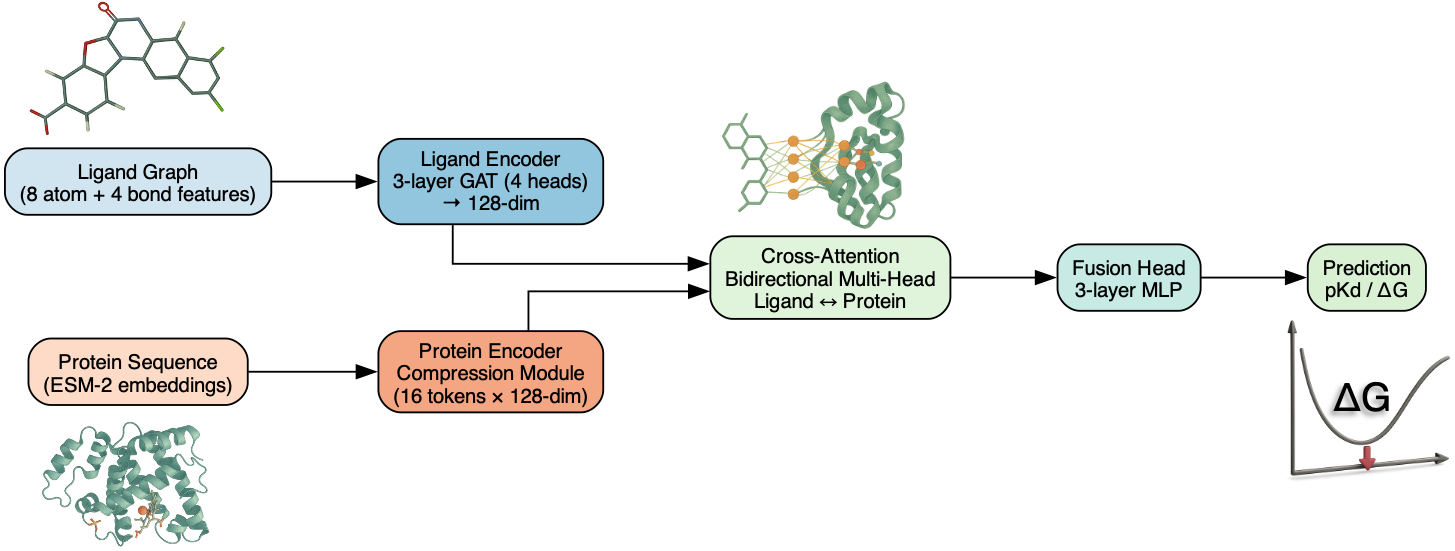
How does MolXProt works
Ligand Encoder (GNN):
- We employ Graph Attention Networks (GAT) with multiple attention heads.
- Input features: 8 atom-features + 4 bond-features for each ligand graph.
Protein Encoder (Language Model Compression):
- We employ pretrained embeddings from the ESM-2 protein language model.
- We further compress the protein representations via multi-head attention to make both representations compatible.
Cross-Attention Layer:
- We use a bidirectional multi-head attention to link ligand tokens and protein tokens.
- This effectively enables the model to learn residue-atom interactions, that is, which amino acids in the protein are most involved in the ligand interaction.
Fusion + Prediction Head:
- Concatenates the attended ligand & protein representations.
- A MLP (Multi-Layer Perceptron) that outputs the predicted binding affinity in pKd or ΔG (kcal/mol).
The data
- Data drawn from public sources (e.g., ChEMBL, BindingDB, Davis).
- Mixed datasets are constructed for robustness.
- Binding ranges: pKd from ~2.1 to ~14.8; ΔG from about -20.2 to -2.9 kcal/mol.
- Standard 70 % train / 10 % validation / 20 % test split.
Results worth noting
On the ΔG (kcal/mol) scale:
| Split | R² | MAE | RMSE |
|---|---|---|---|
| Train | 0.46 | 1.27 | 1.67 |
| Val | 0.42 | 1.31 | 1.71 |
| Test | 0.44 | 1.30 | 1.71 |
In terms of chemical accuracy:
- ~50 % of predictions lie within ±1.0 kcal/mol
- ~80 % lie within ±2.0 kcal/mol
What we learned
- Interpretability in action: By mapping attention weights, we found that the model tends to highlight known binding-pocket residues (for example in benchmarks such as CDK2–Staurosporine and DHFR–Methotrexate). On the other side, hydrogen-bond networks are seem to be underepresented, opening the way to future works incorporating geometry-aware supervision or hierarchical attention.
- Latent space structure: By visualising the latent space representations (via PCA/t-SNE), it reveals a smooth and continuous manifold of binding affinities, with no chemistry pockets dominating or overfitting for specific families.
- Bias in predictions: We found that strong binders tend to be underpredicted, while weak binders tend to be overpredicted. Appears to be linked to dataset imbalance and experimental noise.
- Correcting bias: A simple post-hoc isotonic regression correction can partially reduce the calibration bias, a cheap but effective fix.
Why it stands out
- Fusion of modalities: We combined distinct representations for proteins and ligands via a novel and lightweight architecture.
- Interpretability: The attention mechanism enables molecular insights into what the model looks at during training and inference, allowing us to connect outputs to real chemistry/biology.
- Efficiency: Despite handling large datasets, the model remains lean (approximatelly 318k parameters), being compatible with modest hardware.
- Novel pathways for improvement: The work highlights clear improvement steps, such as better capturing hydrogen-bond networks via geometry-informed tokenisation, or attention hierarchies.
Looking ahead
- Potentially extend dataset coverage and include features derived from MD/DFT.
- Push model capacity to its limits by taking advantage of HPC centers.
- Open source the code and enable community benchmarking once the manuscript is published.
Final word
We believe that with MolXProt we’re taking a meaningful step toward interpretable and scalable binding affinity prediction, where one doesn’t need to sacrifice speed for insight, while also offering new ways to peek into how ligands and proteins really interact. We expect the pre-print to be available soon, stay tuned!